

Nature Communications | 深度挖掘人类肠道宏基因组的古菌病毒
北京时间12月30日,中国科学院深圳先进技术研究院合成生物学研究所马迎飞团队在国际学术期刊《自然-通讯》(nature communications,IF=17.69)上发表了题为"Metagenomic analysis reveals unexplored diversity of archaeal virome in the human gut"的文章。该工作首次对来自人类肠道宏基因组的古菌病毒进行了深入挖掘及全面分析,揭示了人类肠道中古菌病毒组的多样性,为人类肠道古菌病毒组的研究提供了前所未有的一瞥。

文章上线截图
人类微生物组的组成和功能与宿主的健康密切相关。除了细菌外,肠道微生物群中的非细菌成员(古菌、真菌和病毒)也在微生物群落的动态演替以及人类的生理、免疫、疾病等方面发挥着重要作用。古菌是人体的共生微生物之一,它们曾在肠道,口腔和皮肤等部位被检测到,与人类疾病密切相关。与细菌相比,古菌在人体内的丰度相对较低,而且大多不可培养,因此与人类相关的古菌经常被忽视。病毒控制着微生物群落的组成和代谢,侵染古菌的病毒在基因组序列和病毒粒子结构方面具有很高的多样性。迄今为止,只有少数研究报道人类肠道内存在古菌病毒,因此,与人类相关的古菌病毒仍然神秘。随着下一代测序技术的发展以及海量数据的产生,基于宏基因组学的方法有助于我们对人类的古菌组和古菌病毒组进行广泛的研究。本文以人类肠道宏基因组数据为研究对象,利用生物信息学的方法,对古菌病毒进行深度挖掘,探究它们的多样性及潜在功能。
人类肠道古菌病毒组的多样性
研究表明约有90%古菌基因组包含CRISPR结构
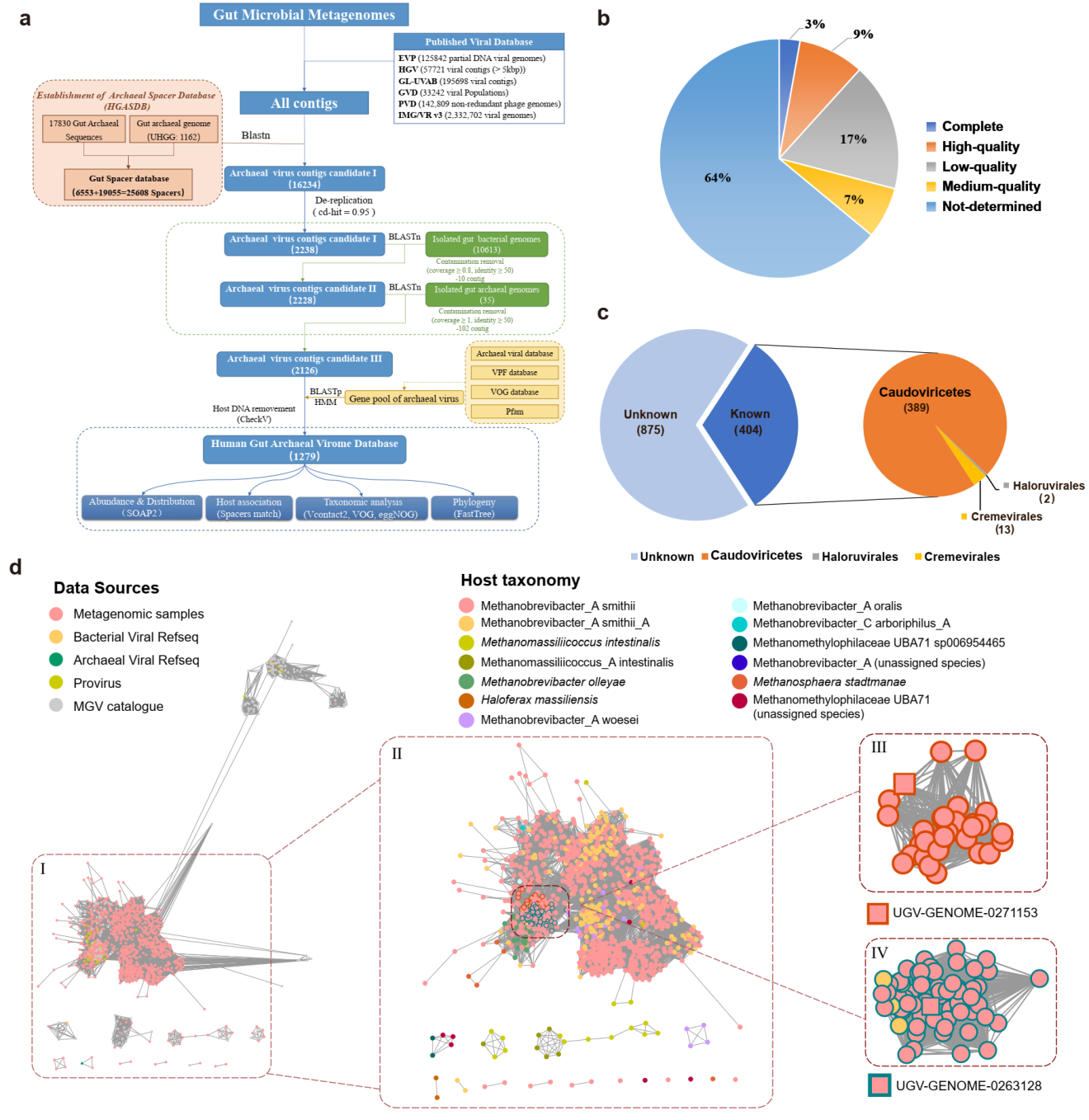
图 1 肠道古菌病毒的鉴定
古菌病毒广泛分布于人类肠道中
为了了解肠道古菌病毒在全球人群中的分布情况,团队对1279条古菌病毒contigs在人类肠道样品中的丰度情况进行了估计,主坐标分析 (PCoA) 和Anosim分析的结果显示:肠道古菌病毒群落的组成在不同性别 (ANOSIM, r = 0.004, p = 0.306) 和不同BMI指数 (ANOSIM, r = 0.006, p = 0.201) 的人群中并无显著性差异。然而,当以国家作为界限分析时,我们观察到这些古菌病毒在各地表现出不同的多样性(图2a)。
团队进一步调查了这些古菌病毒在所有肠道样品中的检出率,共有7种古菌病毒在人群中的检出率>10%,虽然这些病毒被归类到了7个不同的病毒簇(VCs)中,但它们的宿主均为Methanobrevibacter_A smithii (M.smithii)。就地理分布而言,这些古菌病毒在非洲人群中的检出率较低,但在亚洲、欧洲和美洲人群中则具有相对较高检出率 (图2b)。在人类肠道中检出率高于1%的古菌病毒共有712种,病毒序列IMG|UGV-GENOME-0271153在人群中具有最高检出率 (72.2%),该序列长度为40.51 kb,被评估为中等质量的基因组,侵染M.smithii古菌。该病毒基因组编码46个基因,其中8个基因被预测为有尾病毒的功能基因 (图2c)。此外,团队从人类肠道样品中共鉴定出13种smacoviruses,它们的长度分布在2.0~2.5kbp之间。这些病毒与古菌基因组上7个不同spacer序列相匹配,其宿主为Methanomassiliicoccus intestinalis 或Methanomassiliicoccus_A intestinalis古菌。与亚洲和美洲人群相比,smacovirus在非洲和欧洲人群中具有更高检出率 (图2d)。
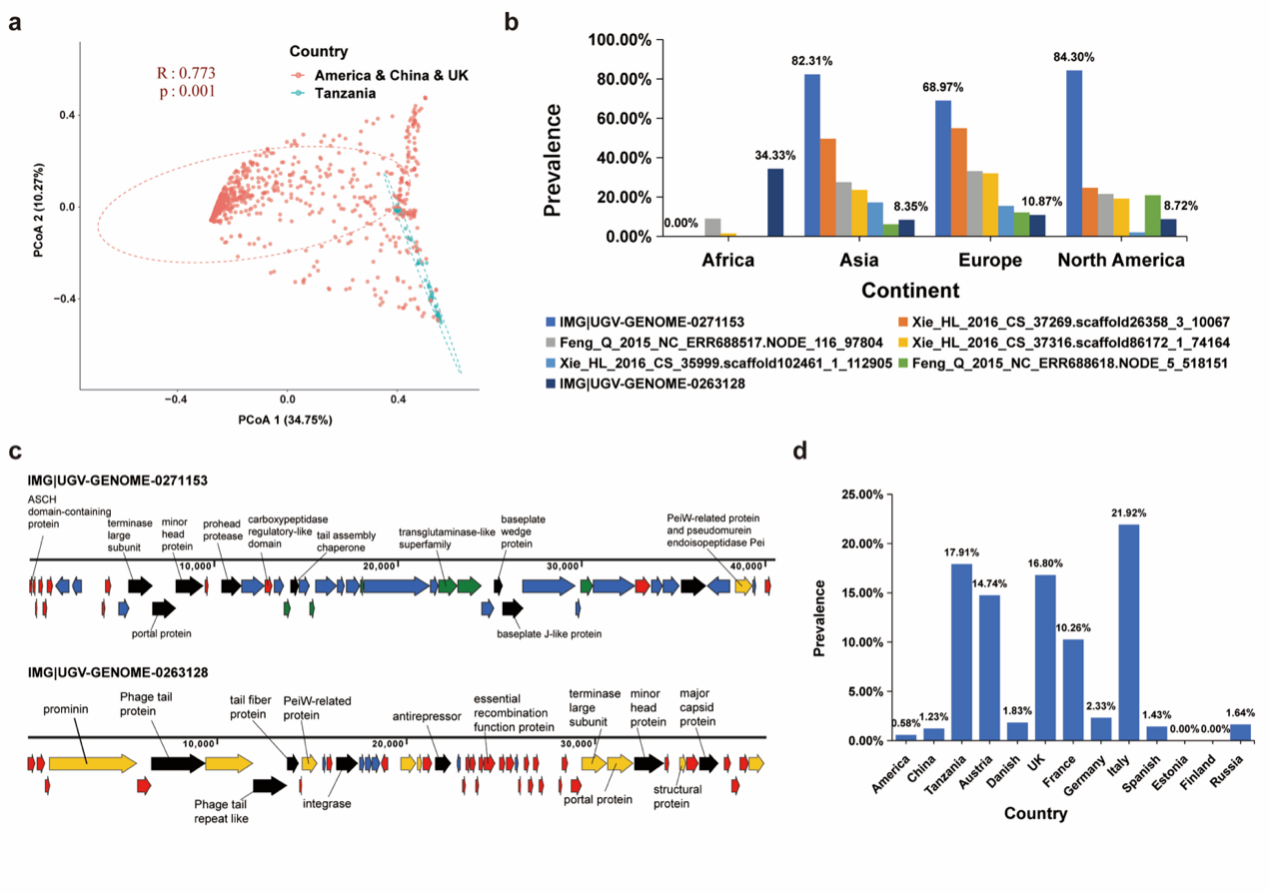
图2 肠道古菌病毒的全球分布
M.smithii古菌的病毒在人类肠道古菌组中占主导地位
病毒能够以多种方式影响生态系统及微生物进化,例如部分噬菌体能够通过裂解宿主细胞改变微生物的群落结构和生态功能;一些噬菌体能够劫持其宿主的代谢机制,改变宿主细胞代谢产物浓度;噬菌体也可以作为宿主之间水平基因转移的载体
此外,侵染M.smithii的病毒是人类肠道古菌病毒的一个主要分支,为了进一步探索有尾古菌病毒的多样性,团队以末端酶大亚基 (Terl, Large Subunit Terminase) 作为标记构建M.smithii古菌病毒的系统发育树,以此来估计这些病毒的多样性(图3d)。以上基于Terl蛋白的系统发育分析扩展了M.smithii古菌病毒的多样性,并定义了新的谱系。
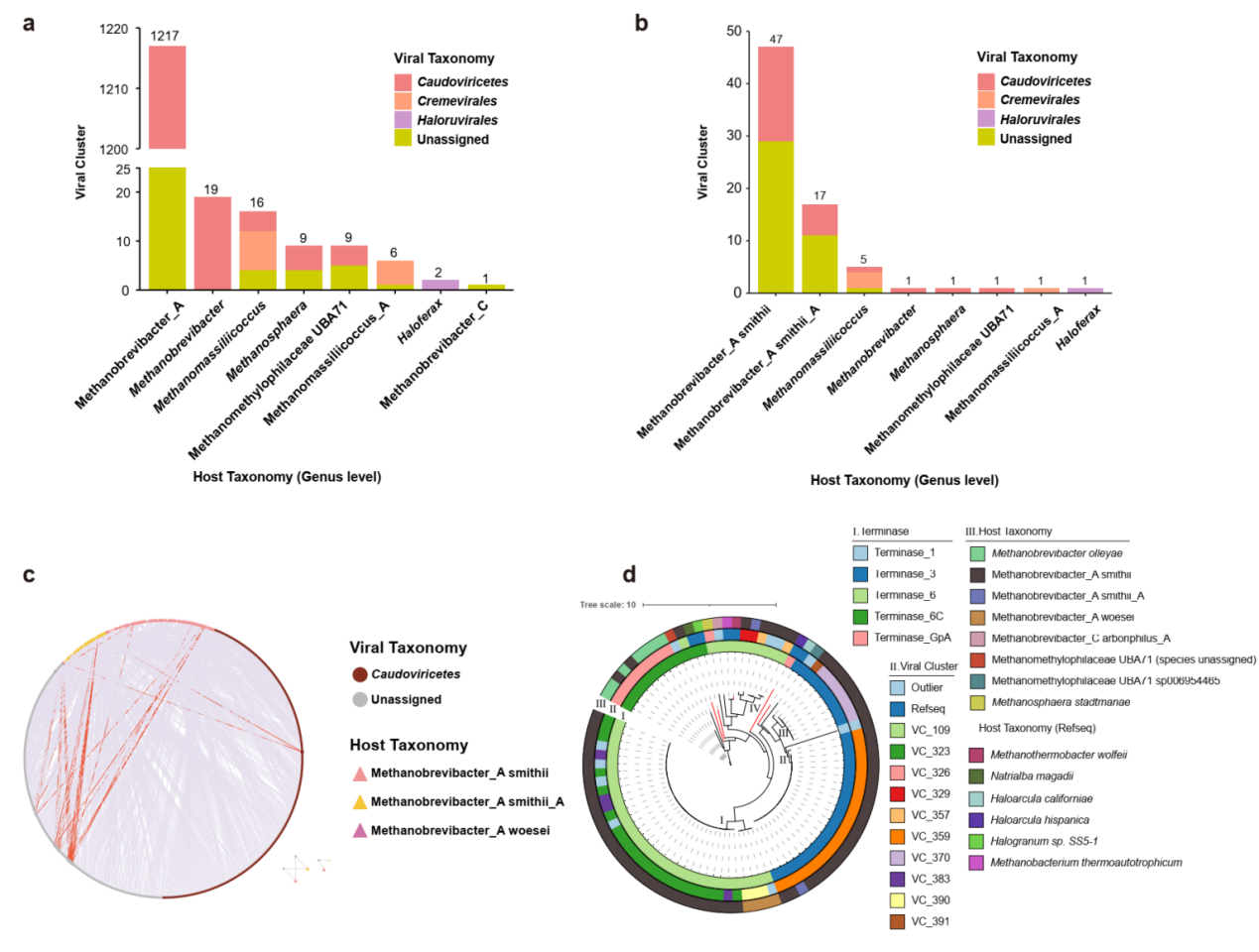
图3 古菌病毒的宿主及宿主范围
古菌病毒基因组编码广泛的蛋白功能
虽然人类肠道中古菌的功能蛋白已被广泛研究,但是对于肠道内古菌病毒蛋白的理解却十分有限。为此,团队从1279条古菌病毒代表序列上共预测出了97,208个编码蛋白的基因,分别仅有 10.8%和 17.4% 的基因能够与pVOG
在所有鉴定出的肠道古菌病毒中,M.smithii古菌病毒的蛋白功能最为多样,共包含了1034种不同功能的蛋白 (只考虑了被指定生物功能的蛋白) ,其中包括一些病毒特有的功能蛋白,例如与结构、包装、裂解、DNA结合/调控和复制相关的蛋白,但其他一些古菌病毒则缺乏这些病毒特有的功能蛋白 (图4b)。此外,团队还对36条完整古菌病毒代表序列的基因组进行了研究 (图4c),其中23条序列携带PeiW(pseudomurein endoisopeptidase)基因,揭示了该基因对于病毒侵染产甲烷古菌的重要性。此外对33种有尾病毒基因组的分析结果表明,与人类肠道中的细菌病毒类似,温和古菌病毒在人类肠道古菌病毒中占据主体地位。
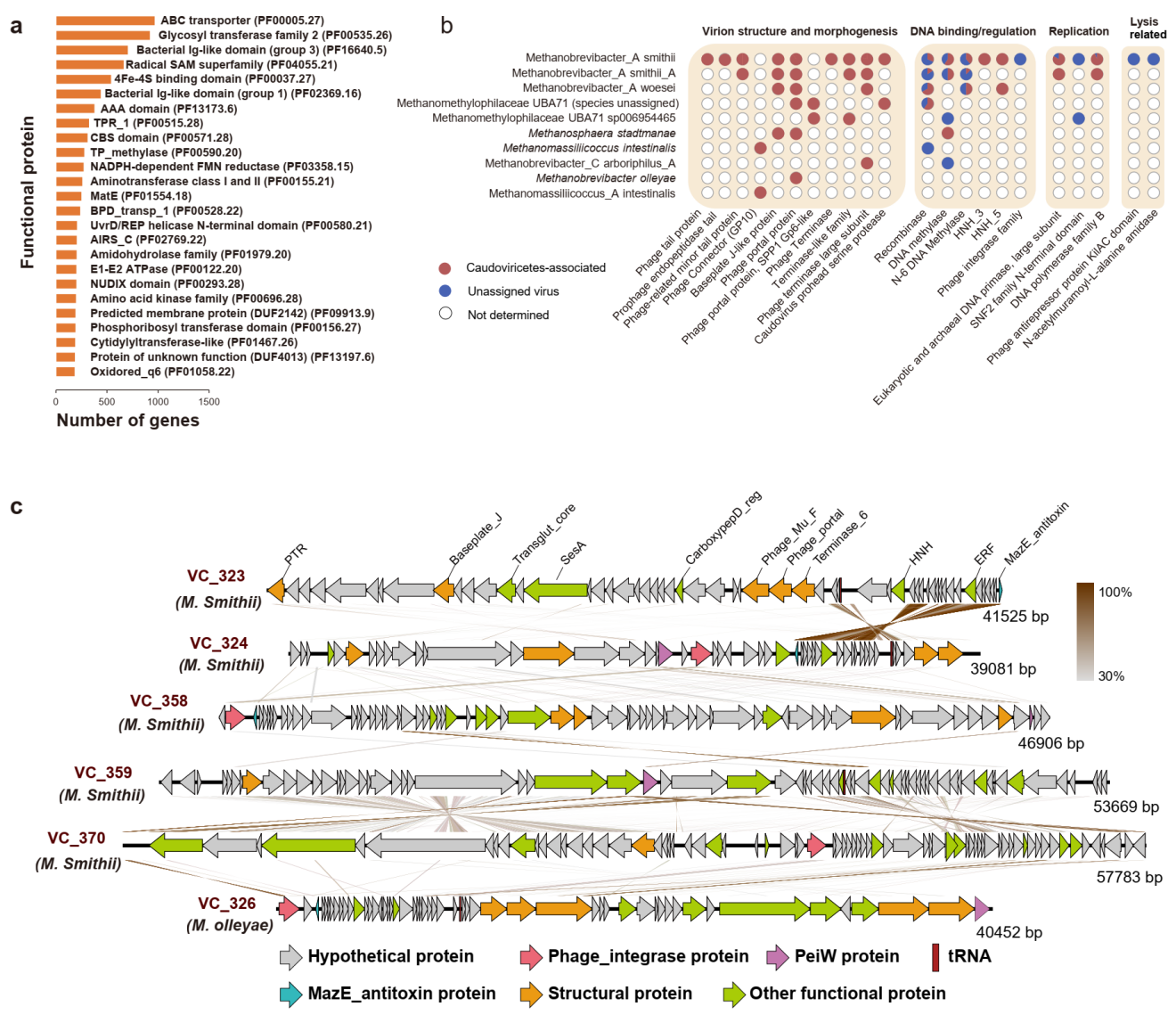
图4 肠道古菌病毒的蛋白功能
在这项研究中,团队对人类肠道中的古菌病毒进行了全面的宏基因组数据挖掘,本研究结果将为深入了解人体肠道内古菌病毒的多样性和蛋白功能提供前所未有的见解,以供人们更好地了解人类肠道生态系统。
中科院深圳先进院马迎飞研究员为该文章的通讯作者,实验室已毕业硕士研究生李冉和助理研究员汪永明为文章共同第一作者。该研究得到了中科院先导B,中科院定量工程生物学重点实验室,广东省合成基因组学重点实验室及深圳合成生物学创新研究院的资助。该研究也得到肠道微生物头部企业未知君(XBIOME)研究团队的支持和帮助。
参考文献
1. Sorek, R., Lawrence, C. M. & Wiedenheft, B. CRISPR-mediated adaptive immune systems in bacteria and archaea. Annual Review of Biochemistry vol. 82 Preprint at https://doi.org/10.1146/annurev-biochem-072911-172315 (2013).
2. Ignacio-Espinoza, J. C. & Sullivan, M. B. Phylogenomics of T4 cyanophages: Lateral gene transfer in the ‘core’ and origins of host genes. Environ Microbiol 14, (2012).
3. Grazziotin, A. L., Koonin, E. v. & Kristensen, D. M. Prokaryotic Virus Orthologous Groups (pVOGs): A resource for comparative genomics and protein family annotation. Nucleic Acids Res (2017) doi:10.1093/nar/gkw975.
4. Terzian, P. et al. PHROG: Families of prokaryotic virus proteins clustered using remote homology. NAR Genom Bioinform 3, (2021).
