

Nature Communications | 数据驱动的微生物群落定殖抗性预测
2024年3月16日,中国科学院深圳先进技术研究院合成微生物组学研究中心、深圳合成生物学创新研究院戴磊课题组在Nature子刊 Nature Communications上发表了肠道微生物组的定量生态学最新研究成果,题为《Data-driven prediction of colonization outcomes for complex microbial communities》。该团队提出并验证了针对微生物组的数据驱动研究范式,用微生物群落的物种组成来准确预测外源物种的定殖结果。深圳先进院的吴璐博士和哈佛大学医学院的王旭文博士为共同第一作者,戴磊研究员和哈佛大学医学院的刘洋彧副教授为文章共同通讯作者。

文章链接:https://www.nature.com/articles/s41467-024-46766-y
人体肠道微生物组在抵抗致病菌定殖过程中发挥着重要作用。这一现象被称为定殖抗性,指的是微生物群落对于外源物种的抵抗能力。研究发现,肠道菌群的物种组成和定殖抗性都呈现出显著的个体差异。针对特定个体的肠道微生物组,全面解析物种之间的互作网络是十分困难的,是否有可能准确预测外源物种的定殖结果?定殖抗性是否由关键物种决定?在这项工作中,研究团队提出并验证了数据驱动的研究范式可以用于预测和理解复杂微生物群落的功能,进而可以通过引入关键物种来实现群落功能的精准调控。
首先,研究团队通过广义Lotka-Volterra模型的解析推导和计算模拟,证明了在训练数据量与微生物群落物种多样性相当的情况下,随机森林、神经常微分方程等机器学习模型可以基于肠道菌群的物种组成准确预测外源物种的定殖结果和稳态丰度(图1)。
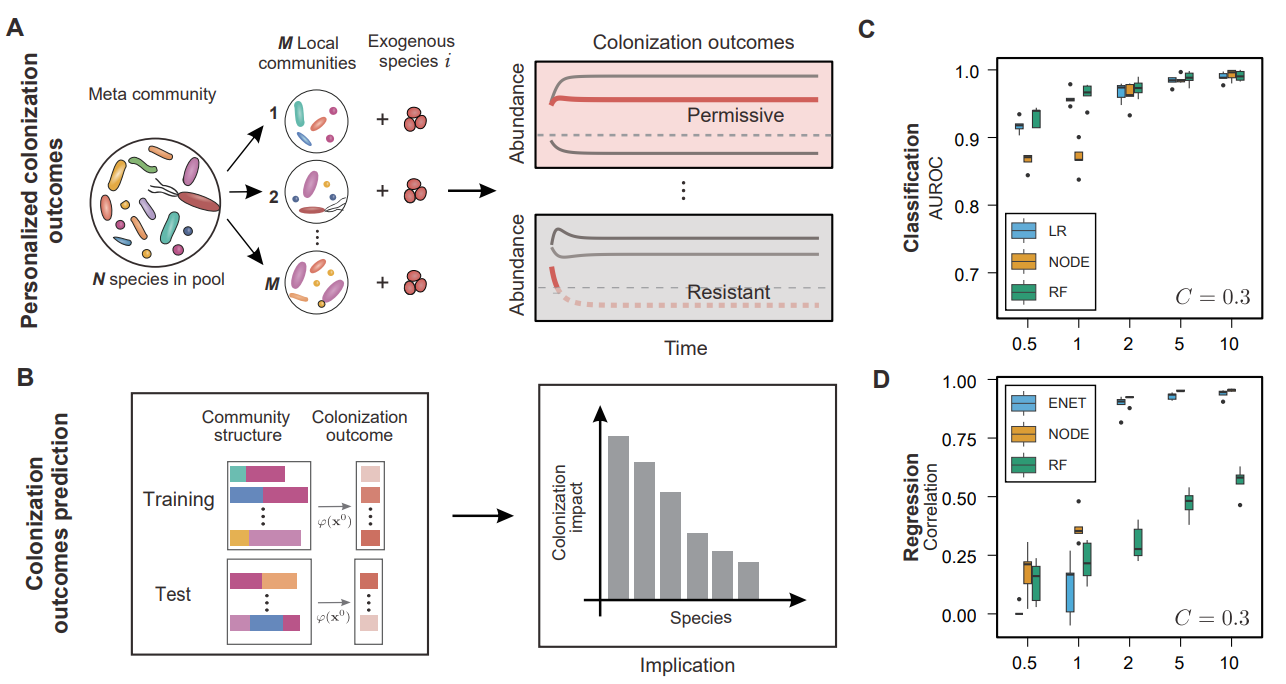
图1. 通过采样不同物种组成的微生物群落作为训练数据,机器学习模型可以准确预测外源物种的定殖结果。
然后,在人体肠道微生物群落的体外培养体系中,研究团队用致病菌Enterococcus faecium和益生菌Akkermansia muciniphila作为代表性物种,在约300个不同物种组成的人体肠道微生物群落中进行了大规模的定殖实验(图2)。实验结果发现,外源物种的定殖结果在不同个体之间存在显著差异。在能够成功定殖的群落中,其稳定丰度也可能存在两个数量级以上的差别。此外,经过抗生素处理后,肠道菌群的物种多样性和定殖抗性显著降低,与之前的经验性观测和理论研究相吻合。对于两个不同物种的定殖实验数据集,机器学习模型均可以基于肠道菌群的初始物种组成来准确预测定殖结果。

图2. 对于来自不同个体的肠道菌群,开展外源物种的定殖实验并对实验结果进行预测。
最后,研究团队通过机器学习模型,开展移除特定物种的假想实验,进一步理解定殖抗性的生态学机制(图3)。模型推断,大多数的共生菌对外源物种有较弱的拮抗作用,这与定殖抗性在复杂群落中的涌现特征相符合。此外,发现某些关键物种的存在可以显著提高定殖抗性,例如Enterococcus faecalis对于Enterococcus faecium的定殖有非常明显的抑制作用,并得到了实验验证。这一结果表明,数据驱动的研究方法可以推断定殖抗性的关键物种,进而指导微生物组的精准调控。通过调控微生物群落的组成来防止致病菌的定殖或者促进益生菌的定殖,在人体健康、农业生产中都有着重要的应用。
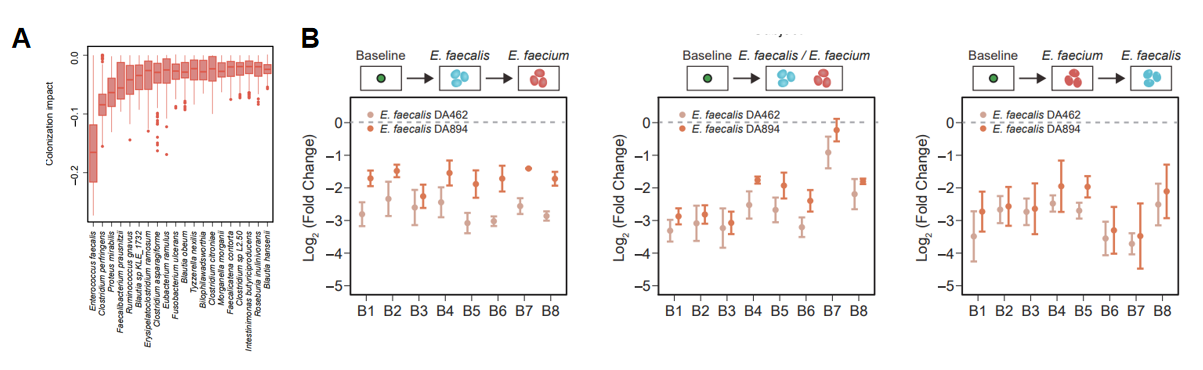
图3. 推断和验证决定肠道菌群定殖抗性的关键物种。
论文特别致谢了深圳合成生物研究重大科技基础设施,其DNA提取自动化平台保证了实验的准确性和可重复性。该工作得到了国家重点研发计划项目(No.2019YFA0906700)、国家自然科学基金(No.31971513, No.32100089)、广东省自然科学基金(No. 2022A1515011513),深圳市微生物药物智能制造重点实验室 (ZDSYS20210623091810032)和深圳合成生物创新研究院的资助。
